easyunfold Documentation#
![]()
![]()
![]()



This package is intended for obtaining the effective band structure of a supercell for a certain k-point path of the primitive cell. It was originally based on PyVaspwfc for reading VASP wavefunction outputs, with a notable improvement being that symmetry-breaking is properly accounted for by sampling necessary additional k-points and averaging accordingly. Typical applications of band structure unfolding are the electronic structure analysis of defects, disorder, alloys, surfaces (and more), as illustrated in the example outputs below and docs examples.
Our goal is to implement the band structure unfolding workflow in a robust and user-friendly software package.
For the methodology of supercell band unfolding, see here.
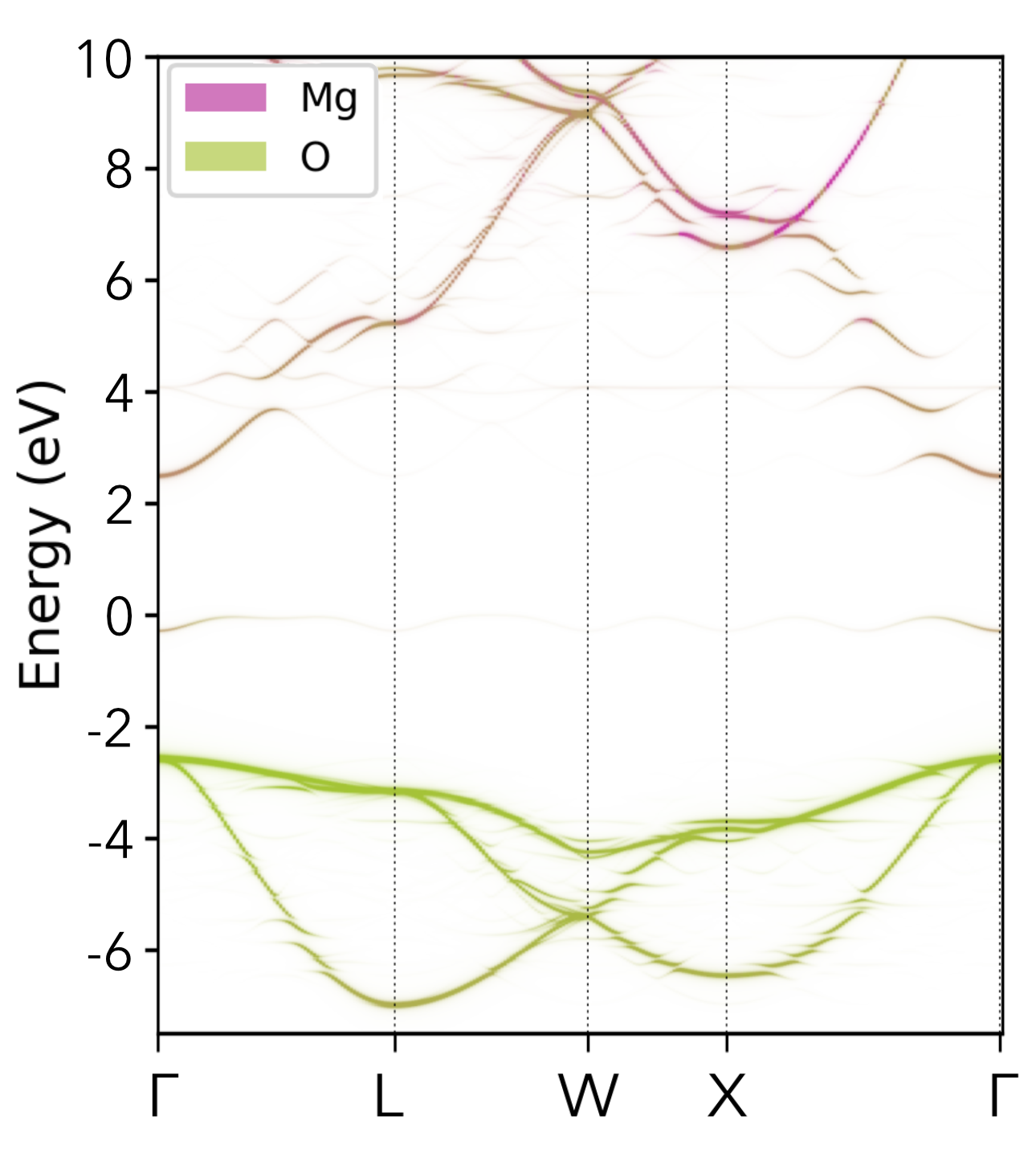
Example Outputs#
Oxygen Vacancy (Vₒ⁰) in MgO |
|
|---|---|
|
|
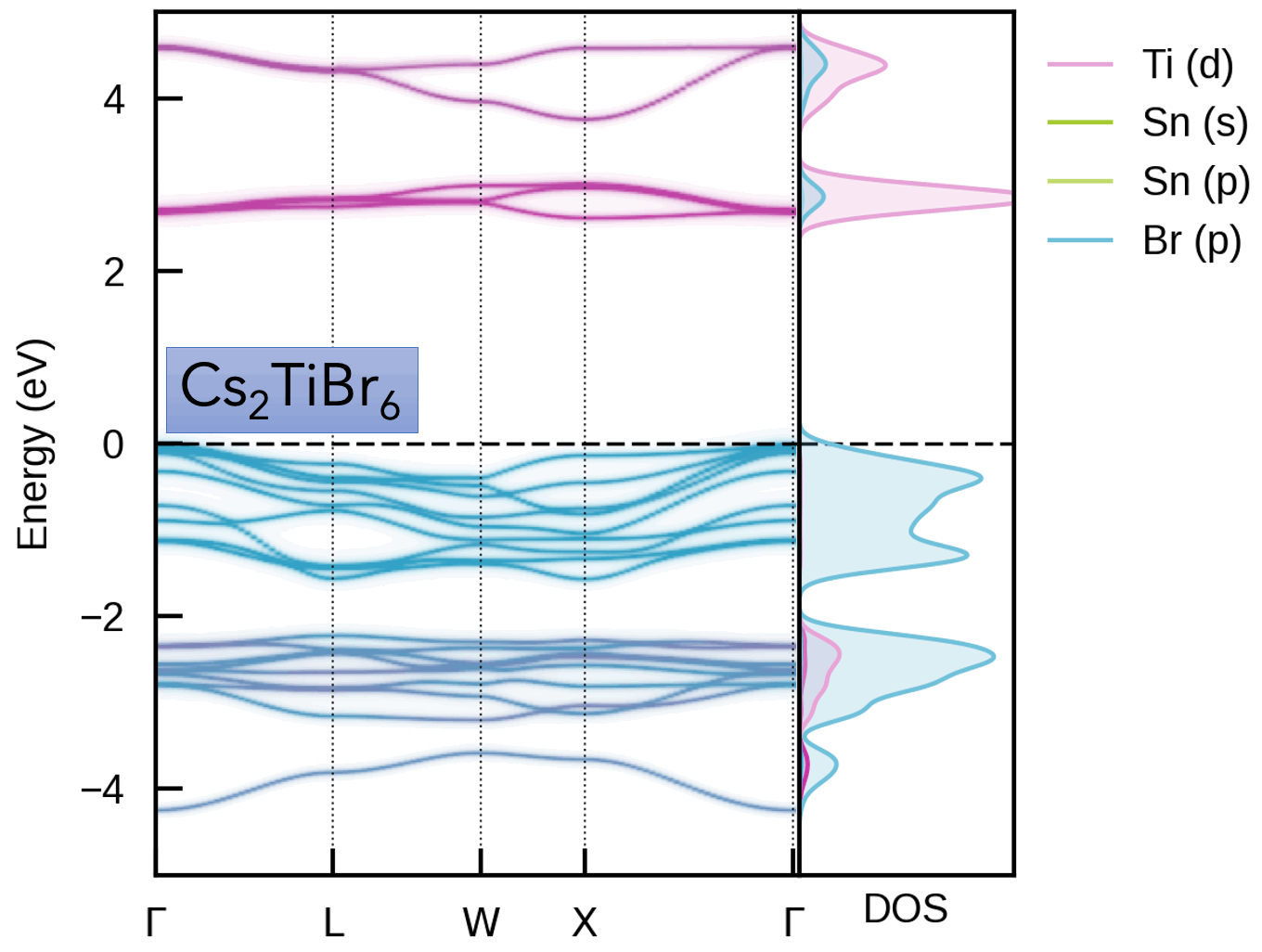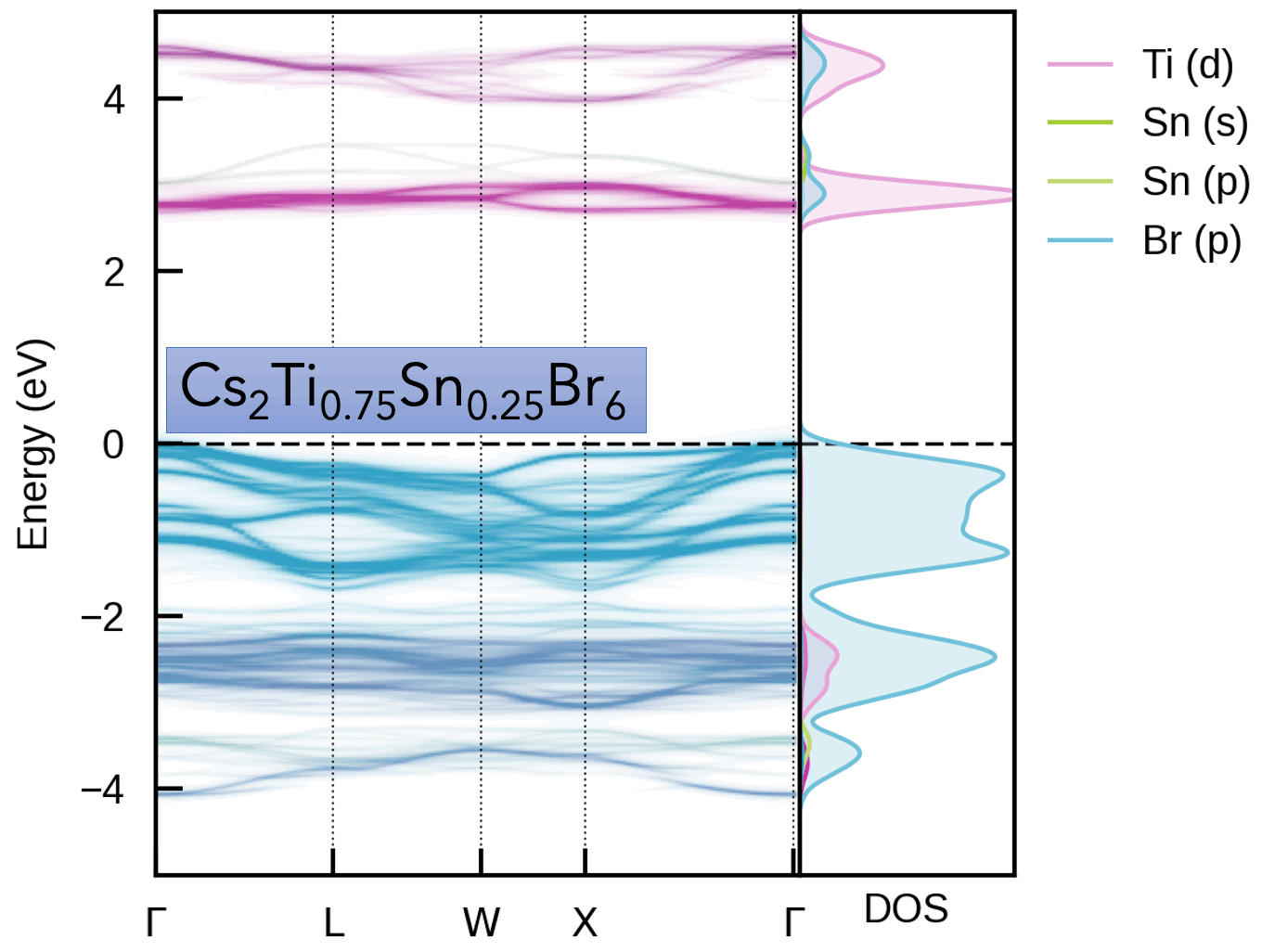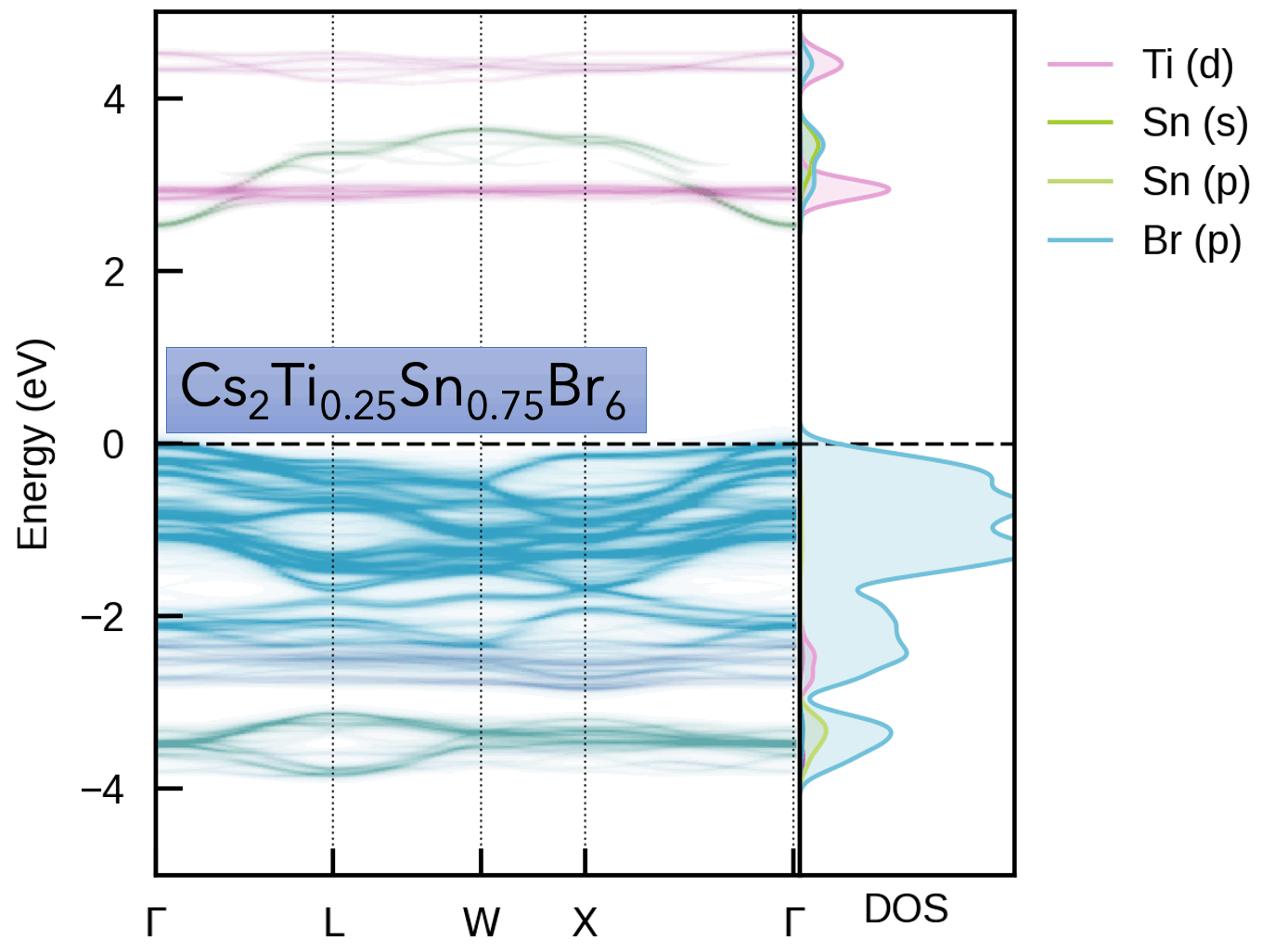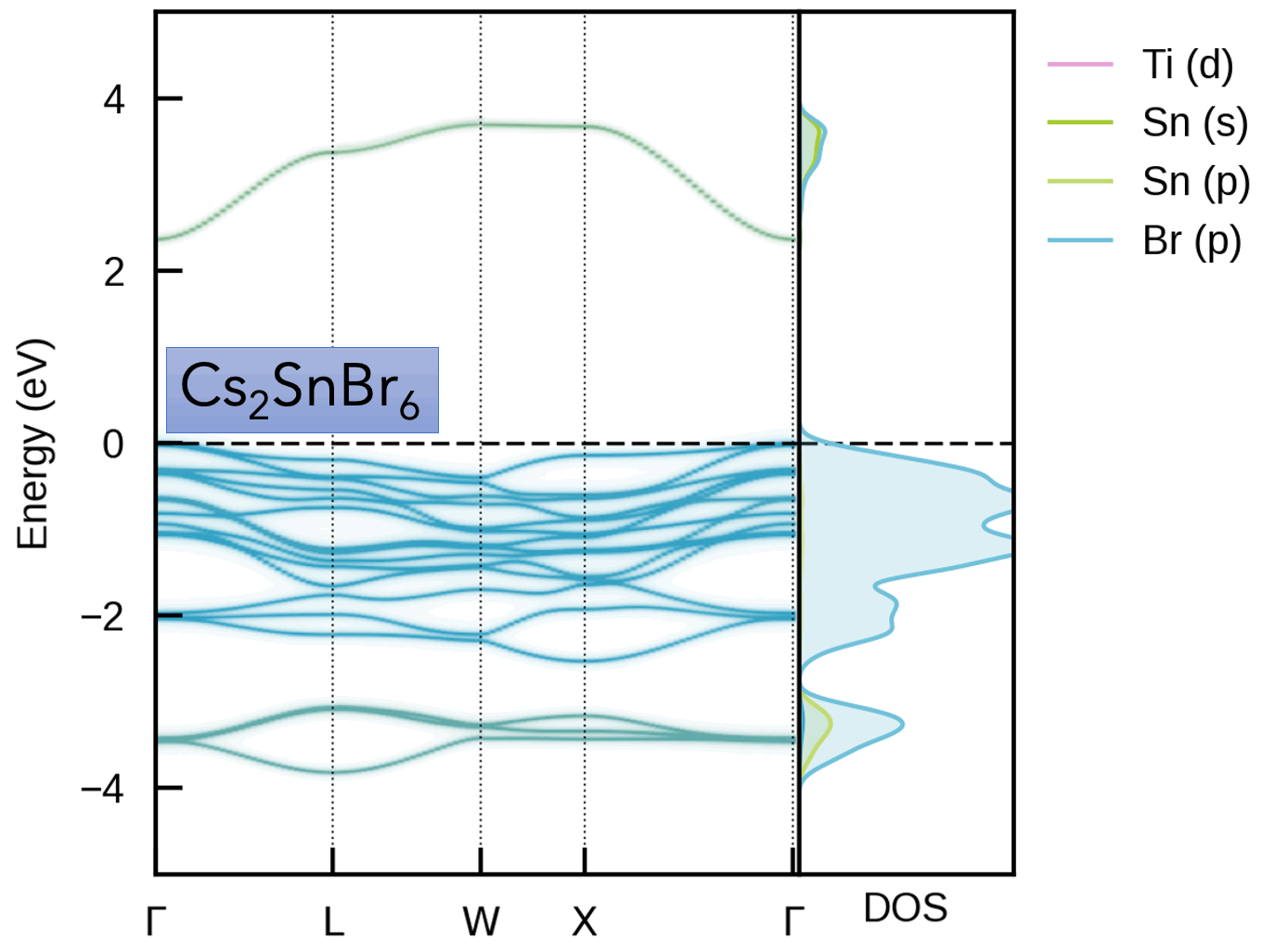
Tip
See the easyunfold YouTube tutorial for a quick overview of the theory of band structure unfolding, and a walkthrough of the calculation & analysis workflow with easyunfold.
Usage#
To generate an unfolded band structure, one typically needs to perform the following steps:
Create a primitive unit cell, and generate a band structure k-point path corresponding to this primitive cell.
Create a supercell (e.g. disordered, defective, surface slab etc.), and obtain its optimised structure.
Generate a series of k-points for the supercell to be calculated.
Perform a band structure calculation with the supercell, and save its wavefunction output to file.
Post-process the supercell wavefunction to obtain the unfolded band structure in the k-point path of the primitive unit cell.
These generation and analysis steps are automated in easyunfold, with only the primitive unit cell and
supercell structures required as inputs from the user.
Typically, the supercell comprises some form of symmetry-breaking relative to the primitive cell, such as defects, disorder (e.g. special quasi-random structures (SQS) for site disorder – other forms of disorder such as magnetic, dynamic/vibrational, polar, elastic etc. also possible), or a surface/interface slab. In all cases, the supercell symmetry is lowered compared to the pristine primitive cell. Hence, for a given k-point path in the primitive cell Brillouin Zone, additional k-points are required to be sampled for the supercell, and the extracted spectral weights need to be appropriately averaged to obtain the correct effective band structure (EBS). See the docs Theory page and/or JOSS paper for more details.
Citation#
If you use easyunfold in your work, please cite:
B. Zhu, S. R. Kavanagh & D. O. Scanlon, (2024). easyunfold: A Python package for unfolding electronic band structures. Journal of Open Source Software, 9(93), 5974, https://doi.org/10.21105/joss.05974
Studies using easyunfold#
We’ll add papers that use easyunfold to this list as they come out!
V. Posligua et al. Deep Learning Framework for Accurate Prediction and High-Throughput Search of the Thermoelectric Figure of Merit in Skutterudites Journal of Materials Chemistry A 2026
K. Li et al. Resonant Doping in Binary Sb(V)-oxide Sb2O5 for High-Mobility Transparent Conductors ChemRxiv 2026
H.-Y. Chen et al. Electrostatically tunable moiré-mediated Wigner states via interfacial potential engineering in 2D van der Waals heterostructures Nature Communications 2026
Ø. Finnseth et al. Electronic Structure and Resonant Circular Dichroism of La0.7Sr0.3MnO3 from Soft X-ray Angle-Resolved Photoemission arXiv 2026
R. Claes et al. Screening ASb2O6 (A = Mg, Ca, Sr, Ba, Cd) for High Performance Transparent Conducting Oxides ChemRxiv 2025
I. Caro-Campos et al. Phonon Scattering by Local Off‐Centering in Diamond‐Like Cu2−XAgxIn2Se4 Chalcopyrites: High Carrier Mobility and Ultralow Thermal Conductivity Small Structures 2026
J. M. Domínguez-Vázquez et al. Enhanced thermoelectric performance in Fe2V0.8W0.2Al thin films: synergistic effects of chemical ordering and tungsten substitution Journal of Materials Chemistry A 2026
H. Li et al. CuI-enhanced thermoelectric performance in GeTe by synchronous modulation of hole concentration and thermal conductivity Matter 2026
A. Pike et al. Alloying to tune the bandgap of the AM2Pn2 Zintl compounds Journal of Physics: Energy 2026
M. Wróblewska et al. Continuous Alloying between Rocksalt and Half-Heusler Structures Drives Metal–Semiconductor Transition in ErNixSb Chemistry of Materials 2026
K. V. Sopiha et al. Functional off-stoichiometry in Cu(In,Ga)Se2. Part II: electronic properties in a wide range of compositions Journal of Materials Chemistry A 2026
Y.-S. Liao et al. Moiré-Induced Electronic Reconstruction in van der Waals Heterobilayer PtSe2/PtTe2 ACS Nano 2026
J. J. Plata et al. High-Entropy Skutterudites as Thermoelectrics: Potential Synthesizability, Enhanced Stability, and Band Convergence via the Cocktail Effect PRX Energy 2026
R. E. Philip et al. Disorder Mediated Fully Compensated Ferrimagnetic Spin-Gapless Semiconducting Behavior in Cr3Al Heusler Alloy Advanced Functional Materials 2026
P. Russell et al. Computational prediction of Y-doped Cd2Sb2O7 as a competitive Sb-based n-type Transparent Conducting Oxide ChemRxiv 2025
P. Kumar et al. Theoretical insights into the h-NbN monolayer for selective and moisture-resistant gas sensing: an ab initio study Electronic Structure 2025
M. Sigl et al. Solution-based synthesis of nanocrystalline KBiS2 films at low temperatures and study of photoinduced charge generation Journal of Materials Chemistry C 2025
Y. Yin et al. Halogen Chains with One-Dimensional Semi-Metallic Electronic Structure and Peierls Physics in Polymorphs of Na4X5 (X = I, Br, Cl) Compounds arXiv 2025
S. Husremović et al. Local interface effects modulate global charge order and optical properties of 1T-TaS2/1H-WSe2 heterostructures ACS Nano 2025
L. Richarz et al. Ferroelectric domain walls for environmental sensors ACS Applied Materials & Interfaces 2025
J. M. Domínguez-Vázquez et al. Thermoelectric performance boost by chemical order in epitaxial L21 (100) and (110) oriented undoped Fe2VAl thin films: an experimental and theoretical study Journal of Materials Chemistry A 2025
J. Tu et al. Giant switchable ferroelectric photovoltage in double-perovskite epitaxial films through chemical negative strain Science Advances 2025
L. F. Leon-Pinzon et al. Observation of Pseudogap in Cr1−xYxN magnetic alloy and its impact on the Seebeck coefficient by ab-initio calculations arXiv 2025
C. Zhang & J. Recatala-Gomez & Z. Aabdin & Y. Jiang et al. Direct Joule-Heated Non-Equilibrium Synthesis Enables High Performing Thermoelectrics arXiv 2025
W. Feng et al. Unraveling the role of oxygen vacancies in the electronic and optical properties of κ-Ga2O3 Communications Chemistry 2025
Y. Gong Wang et al. Influence of Vanadium and Chromium Doping on the Thermoelectric Performance of AgSbTe2 Physica Scripta 2025
P. B. Colominas et al. Giant Thermally Induced Band-Gap Renormalization in Anharmonic Silver Chalcohalide Antiperovskites Journal of Materials Chemistry C 2025
E. Moradpur-Tari et al. A correlation-based optimization model to recover lost and distorted data from scanning tunneling microscopy images based on density functional theory Ultramicroscopy 2025
L. Zhang et al. Mg doping point defects in Al0.5Ga0.5N by density functional theory Vacuum 2025
L. Zhang et al. Impurity point defects in Mg doping Al0.5Ga0.5N: A first principles study Computational Materials Science 2025
L. Zhang et al. Study of native point defects in Al0.5Ga0.5N by first principles calculations Computational Materials Science 2024
H. Maleki-Ghaleh et al. Visible Light-Sensitive Sustainable Quantum Dot Crystals of Co/Mg Doped Natural Hydroxyapatite Possessing Antimicrobial Activity and Biocompatibility Small 2024
K. Eggestad, B. A. D. Williamson, D. Meier and S. M. Selbach Mobile Intrinsic Point Defects for Conductive Neutral Domain Walls in LiNbO3 Journal of Materials Chemistry C 2024
Dargahi et al. Synthesis and characterization of zinc-doped hematite nanoparticles for photocatalytic applications and their electronic structure studies by density functional theory Optical Materials 2024
S. M. Liga & S. R. Kavanagh, A. Walsh, D. O. Scanlon and G. Konstantatos Mixed-Cation Vacancy-Ordered Perovskites (Cs2Ti1–xSnxX6; X = I or Br): Low-Temperature Miscibility, Additivity, and Tunable Stability Journal of Physical Chemistry C 2023
Y. T. Huang & S. R. Kavanagh et al. Strong absorption and ultrafast localisation in NaBiS2 nanocrystals with slow charge-carrier recombination Nature Communications 2022
A. T. J. Nicolson et al. Interplay of Static and Dynamic Disorder in the Mixed-Metal Chalcohalide Sn2SbS2I3 Journal of the Americal Chemical Society 2023
Y. Wang & S. R. Kavanagh et al. Cation disorder engineering yields AgBiS2 nanocrystals with enhanced optical absorption for efficient ultrathin solar cells Nature Photonics 2022 (early version)
DFT code support#
At the moment, easyunfold supports VASP and CASTEP, but most of the routines are abstracted from the code specific details. In principle, support for other plane wave DFT code can be added by:
Implementing a subclass of
WaveFunctionthat handles reading the wave function output.Implementing functions for reading/writing k-points.
Adding branches for dispatching based on the
dft_codeattribute of theUnfoldKSetobject in various places within the code.
The Atomic Simulation Environment (ASE) is used by easyunfold for
reading in structures, so structure file IO is natively supported for essentially all public DFT codes.
In fact, ASE can already run band structure calculations using many plane-wave DFT codes. However, reading the plane wave coefficients from calculation outputs is not widely supported yet, which are needed here for band unfolding. Nevertheless, using ASE’s existing IO framework to widen the code support can be a fruitful direction for further development.
Code Compatibility Notes#
Atom-projected band structures are currently only supported for
VASPcalculation outputs.Gamma-only and non-collinear spin calculations are not supported for
CASTEP.
Contributors#
And those who helped in the development:
Bugs reports and feature requests#
Bug reports and feature requests are well come. If you found any bug or missing features please report it on the Issue Tracker.
Seeking support#
If you need support about using the software, please open a ticket with the help wanted label on the Issue Tracker.
Contributing#
Code contributions#
We welcome your help in improving and extending the package with your own contributions. This is managed through GitHub pull requests; for external contributions, we prefer the fork and pull workflow while core developers use branches in the main repository:
First open an Issue to discuss the proposed contribution. This discussion might include how the changes fit
easyunfold’s scope and a general technical approach.Make your own project fork and implement the changes there. Please keep your code style compliant and use the
pre-commithooks.Open a pull request to merge the changes into the main project. A more detailed discussion can take place there before the changes are accepted.
Development#
To develope the package, please clone it on GitHub and install it with the doc and test extras so all dependencies needed for development are installed:
git clone https://github.com/SMTG-Bham/easyunfold
pip install -e "./easyunfold[doc,test]"
To run the tests, simply run the pytest command while in the top-level code repository.
To build the documentation, run the make html command while in the docs directory.
If you want to add new features, please also include a test case to check that they work as expected. For more information please consult pytest’s documentation.
Please note that we use pre-commit hooks for code formatting and linting.
Please install them using pre-commit install so these hooks can run before committing code updates.