Silicon with a displaced atom#
Below is a step-by-step guide for unfolding the electronic structure of a 2x2x2 supercell of
crystalline silicon (Si) which contains a displaced atom, breaking symmetry.
Generate the project file (easyunfold.json) and k-points for supercell calculation#
Note
The files needed for this example are provided in the examples/Si222 folder. This guide assumes the current working directory is located at the root of that folder.
First, generate the supercell k-points:
easyunfold generate Si/POSCAR Si_super_deformed/POSCAR Si/KPOINTS_band
Here, KPOINTS_band is the KPOINTS file corresponding to the band structure path for the primitive
unit cell, which in this case was generated using sumo-kgen (see Step 1
of the tutorial docs page).
Note
In this example we are using GGA DFT, but if we were using hybrid DFT with VASP, we would need to use the --scf-kpoints option with easyunfold generate, as described in the tutorial docs page.
This generates an easyunfold.json file in the current direction containing information about the
unfolding. The output name of this file can be modified with the --out-file commandline argument.
Information stored in this file can be inspected with the easyunfold unfold status command:
$ easyunfold unfold status
Loaded data from easyunfold.json
Primitive cell information:
Space group number: 160
International symbol: R3m
Point group: 3m
Supercell cell information:
Space group number: 227
International symbol: Fd-3m
Point group: m-3m
No. of k points in the primitive cell : 73
No. of supercell k points : 103
No. of primitive cell symmetry operations : 48
No. of supercell symmetry operations : 6
Path in the primitive cell:
\Gamma : 1
L : 21
W : 38
X : 50
\Gamma : 73
Please run the supercell band structure calculation and run `unfold calculate`.
Copy the k-points to the supercell calculation folder:
cp KPOINTS_easyunfold Si_supercell_deformed
Perform the supercell band structure calculation#
Semi-local (GGA) DFT band structure calculations in VASP normally involve two steps. First, a normal single point calculation is performed to obtain the self-consistent (SCF) charge density. Following this, a non-self-consistent calculation is carried out to compute the eigenvalues of the k-points along the defined band structure path.
First, we run our single point SCF supercell calculation, ensuring to use the appropriate converged SCF
k-point mesh for the supercell. The ICHARG = 11 tag must not be set or be commented out in the INCAR
file for this single point calculation:
cd Si_supercell_deformed
cp KPOINTS_scf KPOINTS # SCF kpoint mesh
sed -i 's/^ICHARG = 11/!ICHARG = 11/g' INCAR # comment out ICHARG = 11
mpirun -np 4 vasp_std # run the calculation
Now, with our converged SCF charge density, we run the GGA band structure calculation with ICHARG = 11,
and the k-points mapped to the supercell from the primitive cell path:
sed -i 's/.*ICHARG = 11/ICHARG = 11/g' INCAR # set ICARG = 11
cp KPOINTS_easyunfold KPOINTS # supercell band structure kpoint path
mpirun -np 4 vasp_std # run the calculation
Alternatively, there is a run.sh script in the
examples/Si222/Si_super_deformed
folder that can be used to perform these two steps above.
Perform band unfolding#
Unfold the supercell wave function (WAVECAR) and calculate the spectral weights:
cd ../
easyunfold unfold calculate Si_super_deformed/WAVECAR
Note
If you don’t want to run the VASP calculation by yourself, the calculated WAVECAR and vasprun.xml
for this example can be downloaded using git-lfs:
If git-lfs was not installed when you cloned the repository, install it via:
sudo apt install git-lfs
git lfs install
(or brew install git-lfs on macOS; see here for full instructions).
Then download the files:
git lfs pull -I examples/Si222/Si_super_deformed/WAVECAR,examples/Si222/Si_super_deformed/vasprun.xml --exclude=""
Plot the unfolded band structure:
easyunfold unfold plot
Output:

Spectral function of the unfolded bands.#
Tip
See the NaBiS2 example for tips on
customising and prettifying the unfolded band structure plot. For example, you can use the --intensity 3.5 option to increase the spectral function intensity.
Note the appearance of extra branches compared to the band structure of the primitive cell (below), due to symmetry breaking from the displaced atom.
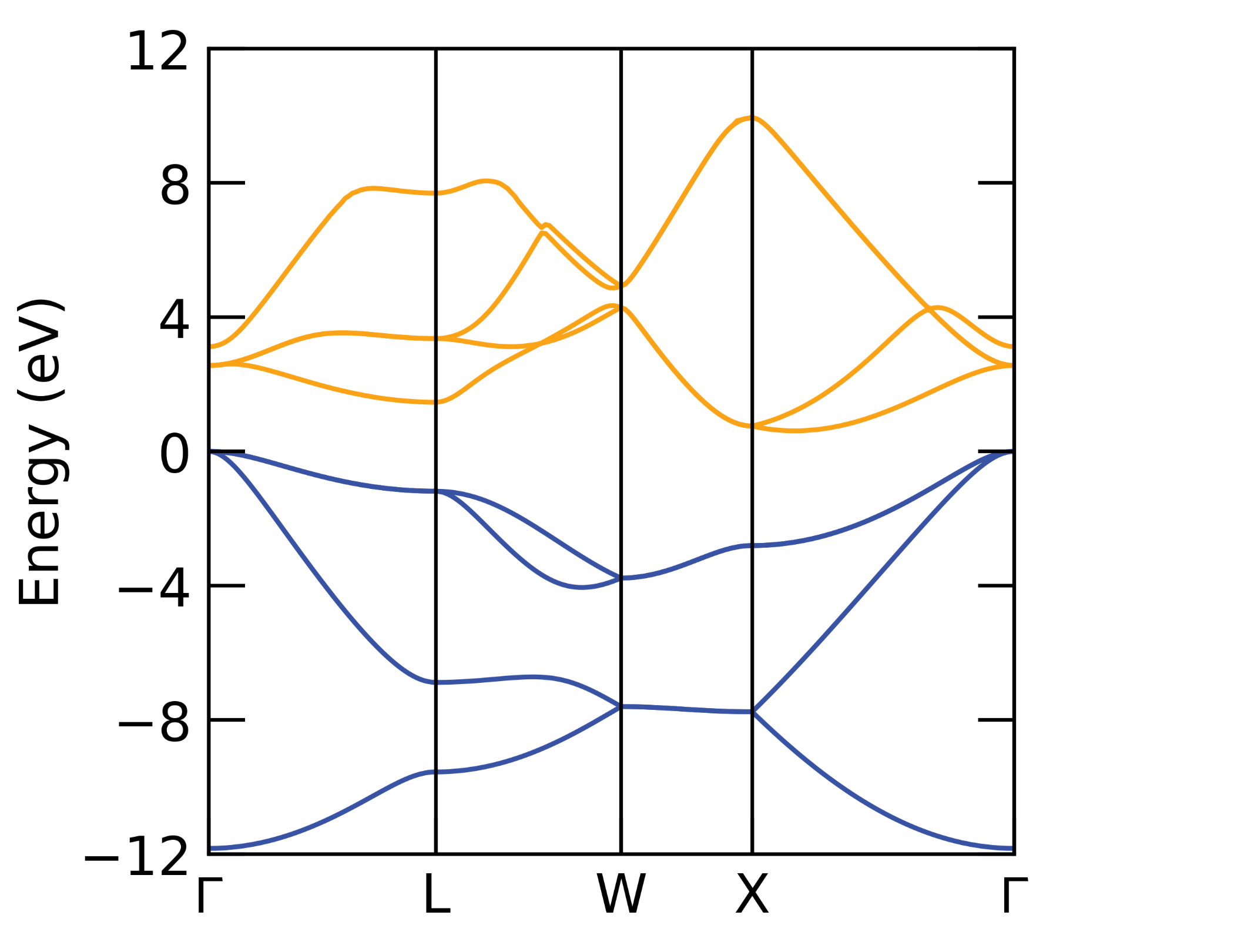
Primitive cell band structure of Si.#
What happens if symmetry is not properly taken into account?#
It is quite common that the supercell has lower symmetry compared to the primitive cell.
By default, easyunfold takes account of such symmetry breaking effect by including
additional k-points that no longer equivalent under the symmetry of the supercell cell.
In this example, we show what happens if we do not include the additional kpoints.
We can create a new unfolding project (json data file) using the following command:
easyunfold generate Si/POSCAR Si_super_deformed/POSCAR Si/KPOINTS_band --no-expand --out-file no-expand.json
Swap the KPOINTS to the new file, non-expanded KPOINTS file:
cp KPOINTS_no-expand Si_super_deformed_no_expand/KPOINTS
cd Si_super_deformed_no_expand
mpirun -np 4 vasp_std
cd ../
easyunfold unfold --data-file no-expand.json calculate Si_super_deformed_no_expand/WAVECAR
easyunfold unfold --data-file no-expand.json plot --out-file unfold_no-expand.png
output:

Spectral function of the unfolded bands without additional kpoints due to reduced symmetry.#
Comparing this plot with the one above, we see that we get spurious band breaking (e.g. along \(\Gamma - L\)) and some branches are missing (near the \(\Gamma\) point).
Nevertheless, by not expanding the k-point paths, fewer supercell k-points need to be calculated:
$ easyunfold unfold --data-file no-expand.json plot --out-file unfold_no-expand.png
Loaded data from no-expand.json
Using a reference energy of 5.284 eV
Unfolded band structure saved to unfold_no-expand.png
$ easyunfold unfold --data-file no-expand.json status
Loaded data from no-expand.json
Supercell cell information:
Space group number: 160
International symbol: R3m
Point group: 3m
Primitive cell information:
Space group number: 227
International symbol: Fd-3m
Point group: m-3m
No. of k points in the primitive cell : 73
No. of supercell k points : 70
No. of primitive cell symmetry operations : 48
No. of supercell symmetry operations : 6
Path in the primitive cell:
\Gamma : 1
L : 21
W : 38
X : 50
\Gamma : 73
Unfolding has been performed - use `unfold plot` to plot the spectral function.
Note that in most cases one would always want to include the additional kpoints to correctly capture the effect of symmetry breaking.
The --no-expand option should be used with care and only when there is no alternative,
for example,
when the expansion gives too many kpoints for very large supercells of special quasi-random structures.
Note
One can always split the workload into multiple calculations with --nk-per-split to fit the computational resources available for individual calculations.